Carzy
2021-07-07 06:07
可以读一下这篇文章 Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study
http://www.nature.com/nbt/jour ... .html
简单的说就是测量基因表达量时,差异不是很大;对于splice junction and variant detection,差异比较大。
RNA-Seq和microarry的吻合性很多人都做过,基本上都说相关性很高。比如这篇文章The concordance between RNA-seq and microarray data depends on chemic优艾设计网_设计LOGOal treatment and transcript abundance
http://www.nature.com/nbt/jour ... .html
要呸一起呸 优艾设计网_Photoshop交流 2021-07-07 06:24
楼上提供了文献,我就根据之前的经验简单说下吧:
RNA-seq建库时一般有个步骤会比较让人纠结,就是拿到cDNA片段的过程,是将mRNA全长直接打断片段化以后再反转录,还是将mRNA全长反转录后再打断比较好,从实际可操作性上来说,一般大部分会选择前者,因为可实践性更好,如果直接针对全长mRNA进行反转录,很难拿到均一的全长cDNA,很多时候最终测到的结果会有相对比较明显的3’端或5‘端偏向性,这对于后期的分析是会有影响的。
可以读一下这篇文章 Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study
http://www.nature.com/nbt/jour ... .html
简单的说就是测量基因表达量时,差异不是很大;对于splice junction and variant detection,差异比较大。
RNA-Seq和microarry的吻合性很多人都做过,基本上都说相关性很高。比如这篇文章The concordance between RNA-seq and microarray data depends on chemic优艾设计网_设计LOGOal treatment and transcript abundance
http://www.nature.com/nbt/jour ... .html
要呸一起呸 优艾设计网_Photoshop交流 2021-07-07 06:24
楼上提供了文献,我就根据之前的经验简单说下吧:
RNA-seq建库时一般有个步骤会比较让人纠结,就是拿到cDNA片段的过程,是将mRNA全长直接打断片段化以后再反转录,还是将mRNA全长反转录后再打断比较好,从实际可操作性上来说,一般大部分会选择前者,因为可实践性更好,如果直接针对全长mRNA进行反转录,很难拿到均一的全长cDNA,很多时候最终测到的结果会有相对比较明显的3’端或5‘端偏向性,这对于后期的分析是会有影响的。

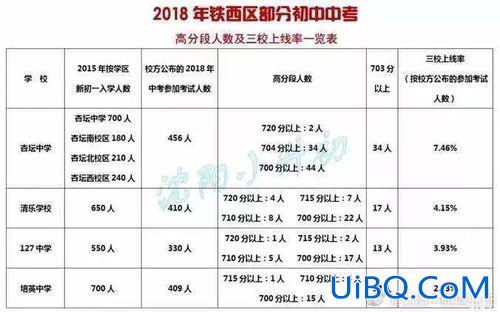



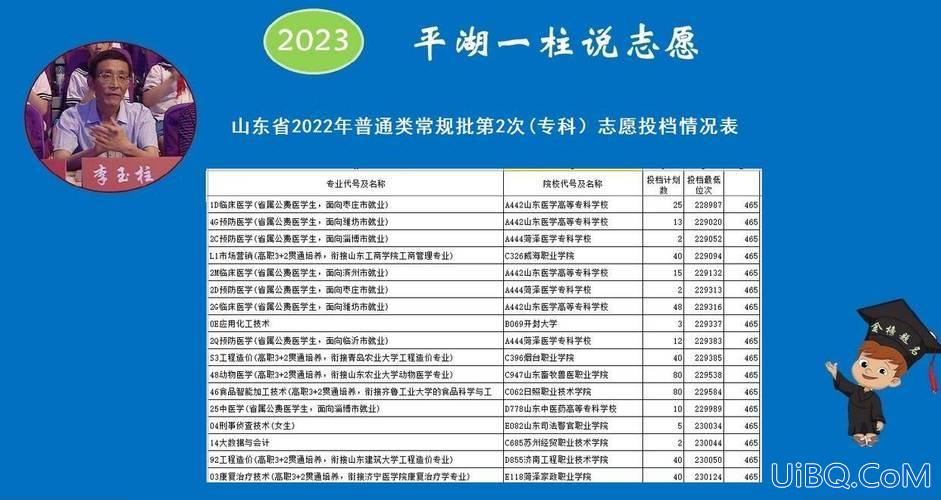


 加载中,请稍侯......
加载中,请稍侯......
精彩评论